Covalent Docking
To model the tethering experiments in silico the ligands should
covalently dock to the flexible side chain of cysteines (e.g., Cys195
of the active site of hTS 1hvy). A covalent docking is possible in GOLD
but it requires a linking atom existing in both, the protein and the
ligand. The linking atom should have an open valence electron (a
hydrogen atom bound to the linking atom of the ligand will be displayed
but not considered in the scoring function). The flexibility of a
protein residue can be specified in GOLD by adding manually a rotamer_lib
block to the gold.conf.
Linking atom
file:///sw/mcm/app/gold/gold_v3.1.1/gold/doc/portable_html/gold_portable-3-049.html
/sw/mcm/app/gold/gold_v3.1.1/gold/rotamer_library.txt
- Protein and ligand have to contain the same linking atom with an
open valence electron.
- A second sulfur atom (SG2)
was added to Cys195 using
pymol.
- The linking atom of the ligands can be either specified by giving
the atom number or by doing a substructure search (substructure.mol2).
The substructure search works quite good for the tioles.
Side chain flexibility
file:///sw/mcm/app/gold/gold_v3.1.1/gold/doc/portable_html/gold_portable-3-023.html
- The flexibility of protein residues is only available for
GOLDScore.
- gold.conf has to be changed manually and
not via the GOLD graphical user interface.
- For each flexible main and side chain a rotamer_lib block must be given.
Problems
- How to combine the flexibility of a protein residue with covalent
docking?
- How to remove parts of the ligand?
R1-S-S-R2 -- R1-S-S*- and R2-S-S*- -- R1-S*-Cys and
R2-S*-Cys
- How to replace parts of the ligand?
R1-S*-H -- R1-S*- and -S*-Cys -- R1-S*-Cys
Problem 1. In GOLD it is not
possible to use the flexibility of a side chain (not shown) to move the
linking atom. The linking atom of the ligand is everytime at the same
position.
(question
to the GOLD support)
A possibility to solve this problem is to use the Cys side chain as a
part of the ligand and attach it to the backbone CA atom of the protein.
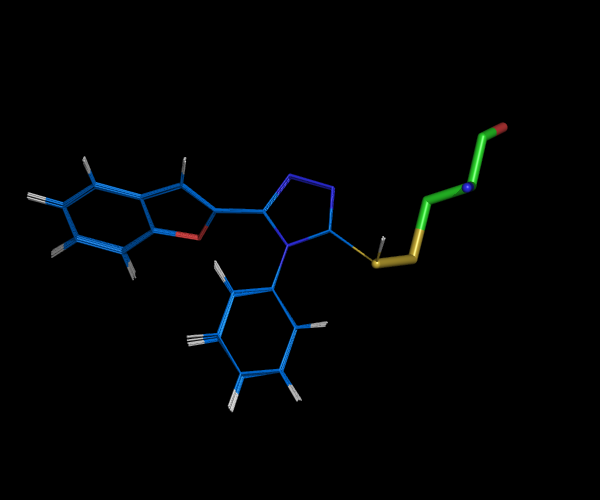
Problem 2 and 3. So far
unsolved!
May be possible with ChemAxon's
Reactor
gold.conf
GOLD CONFIGURATION FILE
generated by gold front end (GOLD v3.1.1)
POPULATION
popsiz = 100
select_pressure = 1.1
n_islands = 5
maxops = 100000
niche_siz = 2
GENETIC OPERATORS
pt_crosswt = 95
allele_mutatewt = 95
migratewt = 10
FLOOD FILL
radius = 15
origin = 0 0 0
do_cavity = 1
floodfill_atom_no = 2703
cavity_file = bmode.mol2
floodfill_center = atom
DATA FILES
protein_datafile = protein.pdb
ligand_data_file tioles.sdf 10
param_file = DEFAULT
set_ligand_atom_types = 1
set_protein_atom_types = 1
directory = .
tordist_file = DEFAULT
make_subdirs = 1
save_lone_pairs = 1
fit_points_file = fit_pts.mol2
read_fitpts = 0
FLAGS
display = 0
internal_ligand_h_bonds = 0
n_ligand_bumps = 0
flip_free_corners = 0
flip_amide_bonds = 0
flip_planar_n = 1
flip_ring_NRR flip_ring_NHR
flip_pyramidal_n = 0
rotate_carboxylic_oh = flip
use_tordist = 1
TERMINATION
early_termination = 0
n_top_solutions = 3
rms_tolerance = 1.5
CONSTRAINTS
force_constraints = 0
COVALENT BONDING
covalent = 1
covalent_protein_atom_no = 2704
covalent_substructure = 1
covalent_substructure_filename
=substructure.mol2
covalent_substructure_atom_no
= 2
covalent_topology = 1
SAVE OPTIONS
save_score_in_file = 1
save_protein_torsions = 1
output_file_format = MACCS
FITNESS FUNCTION SETTINGS
initial_virtual_pt_match_max =
2.5
relative_ligand_energy = 0
score_param_file = DEFAULT
start_vdw_linear_cutoff = 4
rotamer_lib
name cys195
chi1 2699 2698 2700 2699
chi2 2698 2699 2702 2703
chi3 2699 2702 2703 2704
rotamer 0 (30) 62
(11) 180 (17)
rotamer 0 (30)
-177 (10) 65 (15)
rotamer 0 (30)
-177 (10) 180 (11)
rotamer 0 (30)
-67 (10) 180 (12)
rotamer 0 (30)
-65 (9) -65 (10)
end_rotamer_lib
substructure.mol2
#
Creating user name: henricsn
#
Creation time: 11. 03. 2007 11:58
# Modifying
user name: henricsn
#
Modification time: 11. 03. 2007 11:58
#
Program: corina_permanent_Linux2 3.20 0003
11.05.2005
@TRIPOSMOLECULE
ZINC00029124.mol
2 1 0
0 0
SMALL
NO_CHARGES
@TRIPOSATOM
1
C1
-0.0225 1.8097 0.0120
C.3
2
S2
0.0021 -0.0041 0.0020
S.3
@TRIPOSBOND
1 1 2 1
# End of record
(selected75_tioles.sdf)
tioles.sdf
ZINC00029124
3D
Structure written
by MMmdl.
32 35
0 0 1
0 999
V2000
-0.1727 1.2404 0.0779 C
0 0 0 0 0 0
0.9414 1.9464 0.5731 C
0 0 0 0 0 0
2.2175 1.3506 0.5566 C
0 0 0 0 0 0
2.3766 0.0464 0.0443 C
0 0 0 0 0 0
1.2623 -0.6605 -0.4530 C 0
0 0 0 0 0
-0.0125 -0.0620 -0.4351 C 0
0 0 0 0 0
3.6521 -0.5544 0.0342 N
0 0 0 0 0 0
4.5504 -0.4824 -0.9579 C 0
0 0 0 0 0
5.6602 -1.1593 -0.6603 N 0
0 0 0 0 0
5.4488 -1.6948 0.6047 N
0 0 0 0 0 0
4.2248 -1.3090 0.9940 C
0 0 0 0 0 0
3.6643 -1.6947 2.2910 C
0 0 0 0 0 0
2.4045 -1.2630 2.6245 O
0 0 0 0 0 0
2.1257 -1.7613 3.8788 C
0 0 0 0 0 0
0.9515 -1.5794 4.6279 C
0 0 0 0 0 0
0.8652 -2.1779 5.8993 C
0 0 0 0 0 0
1.9457 -2.9398 6.3920 C
0 0 0 0 0 0
3.1163 -3.1087 5.6203 C
0 0 0 0 0 0
3.2306 -2.5189 4.3397 C
0 0 0 0 0 0
4.2123 -2.4607 3.2953 C
0 0 0 0 0 0
4.2821 0.4237 -2.5068 S
0 0 0 0 0 0
-1.1509 1.6982 0.0914 H
0 0 0 0 0 0
0.8170 2.9446 0.9662 H
0 0 0 0 0 0
3.0700 1.8930 0.9388 H
0 0 0 0 0 0
1.3823 -1.6601 -0.8448 H 0
0 0 0 0 0
-0.8676 -0.6020 -0.8142 H 0
0 0 0 0 0
0.1343 -0.9928 4.2350 H
0 0 0 0 0 0
-0.0264 -2.0540 6.4966 H
0 0 0 0 0 0
1.8766 -3.3979 7.3679 H
0 0 0 0 0 0
3.9345 -3.6949 6.0117 H
0 0 0 0 0 0
5.1859 -2.9279 3.3032 H
0 0 0 0 0 0
5.4700 0.1284 -3.0421 H
0 0 0 0 0 0
1 6
2 0 0 0
1 2
1 0 0 0
1 22
1 0 0 0
2 3
2 0 0 0
2 23
1 0 0 0
3 4
1 0 0 0
3 24
1 0 0 0
4 5
2 0 0 0
4 7
1 0 0 0
5 6
1 0 0 0
5 25
1 0 0 0
6 26
1 0 0 0
7 11
1 0 0 0
7 8
1 0 0 0
8 9
2 0 0 0
8 21
1 0 0 0
9 10
1 0 0 0
10 11
2 0 0 0
11 12
1 0 0 0
12 20
2 0 0 0
12 13
1 0 0 0
13 14
1 0 0 0
14 19
2 0 0 0
14 15
1 0 0 0
15 16
2 0 0 0
15 27
1 0 0 0
16 17
1 0 0 0
16 28
1 0 0 0
17 18
2 0 0 0
17 29
1 0 0 0
18 19
1 0 0 0
18 30
1 0 0 0
19 20
1 0 0 0
20 31
1 0 0 0
21 32
1 0 0 0
M END
$$$$
mono1hvy_4tethering_3.pdb
...
ATOM 2698
N CYS 195
7.739 -7.833 8.777 1.00
20.64 N
ATOM 2699
CA CYS 195
7.659 -6.656 7.922 1.00
20.35 C
ATOM 2700
C CYS 195
8.864 -5.802 8.312 1.00
19.16 C
ATOM 2701
O CYS 195
9.350 -4.994 7.521 1.00
20.76 O
ATOM 2702
CB CYS 195
6.347 -5.895 8.163 1.00
19.79 C
ATOM 2703
SG CYS 195
4.869 -6.842 7.681 1.00
20.42 S
ATOM 2704 SG2
CYS 195 4.264
-8.012 9.251 1.00
20.42 S
ATOM 2705 H02
CYS 195 7.106
-7.925 9.558 1.00
20.64 H
ATOM 2706 H03
CYS 195 7.670
-6.917 6.864 1.00
20.35 H
ATOM 2707 H04
CYS 195 6.371
-4.971 7.586 1.00
19.79 H
ATOM 2708 H06
CYS 195 6.275
-5.706 9.234 1.00
19.79 H
...