BD
simulation results
Simulation details
Channeling rate
Electrostatic potentials
at GLN and GLU binding sites
Brownian dynamics simulation
trajectories
Simulation details
Brownian motions of charged and
uncharged probes of different radii (0.1-1.4 A) were simulated in the presence
of rigid protein.
Starting point for simulations
was a 3A vicinity of the geometric center of the following atoms of HisH
active site residues:
SG_CYS_B_84, NE2_HIS_B_178, OE1_GLU_B_180
and OE2_GLU_B_180 (or chains D or F for other copies of HisHF).
End (target) point of simulations
was variable (defined by reaction distance) vicinity of the geometric center
of the following atoms of HisF active site residues:
ND2_ASN/ASP_A_11, OD1_ASP_A_130 and OD2_ASP_A_130
(or chans C or E).
To simulate only the passage
of a substrate through the channel (exclude trajectories reaching the end
point through the solvent), the trajectories were stopped when the substrate
moves further than 20 A from the center of HisHF . This ensures that
no trajectory approaches the end point without passing the channel.
Additional simulations were done with
trajectory stopping distance of 400 A. The rates from these simulations
were 3-4 times larger than the rates when trajectories truncated at 20
A, i.e. there is ca 1 channeling trajectory per 3-4 successfull trajectories.
A few simulations were done with the
different choice of the starting point (NE2 of bound Gln, CD of bound Glu,
active site center defined by PASS) - no significant changes in channeling
rates.
Ammonia has a dipole moment 1.47 D
(comparable to the dipole moment of water molecule,1.855 D). Modeling
it as dipole (as having 2 or 4 charges) did not result in large differences
from neutral probe.
Channeling rate
- approaching the target point to within 14 A (K99 is passed) and 5 A (S101
is passed)
|
Structure
|
chains
|
Comments
|
largest channeling probe (A)
|
channeling rate for a neutral probe
(%)
|
channeling rate for a charged probe
(%)
|
|
|
|
|
|
|
|
aicar
|
AB
|
|
1.1 / 0.8
|
11.3 / 2.7
|
54.4 / 1.2
|
|
CD
|
ligand removed
|
0.9 / 0.8
|
11.3 / 2.1
|
16.9 / 0.3
|
|
EF
|
|
1.2 / 0.8
|
13.0 / 2.9
|
5.8 / 0.2
|
|
|
|
|
|
|
|
gln
|
AB
|
ligand removed
|
0.8 / 0.7
|
11.8 / 2.3
|
10.7 / 1.1
|
|
CD
|
|
0.7 / 0.7
|
11.1 / 2.3
|
6.0 / 0.3
|
|
EF
|
ligand removed
|
0.7 / 0.7
|
10.4 / 2.2
|
7.8 / 1.7
|
|
|
|
|
|
|
|
glut
|
AB
|
ligand removed
|
1.1 / 0.7
|
10.9 / 2.5
|
42.7 / 1.6
|
|
CD
|
ligand removed
|
1.2 / 0.8
|
10.8 / 2.2
|
43.4 / 1.2
|
|
EF
|
|
1.1 / 1.1
|
11.8 / 3.9
|
43.4 / 7.3
|
|
|
|
|
|
|
|
imgp-gln
|
AB
|
ligand removed
|
0.8 / 0.8
|
11.8 / 2.5
|
10.3 / 2.3
|
|
CD
|
|
0.7 / 0.7
|
10.3 / 2.1
|
17.1 / 0.2
|
|
EF
|
ligands removed
|
0.7 / 0.7
|
11.2 / 2.6
|
12.5 / 7.5
|
|
|
|
|
|
|
|
1jvn
|
AA
|
acivicin left
|
1.0 / 0.8
|
29.0 / 10.6
|
-
|
|
BB
|
acivicin left
|
0.8 / 0.7
|
27.2 / 10.9
|
-
|
|
|
|
|
|
|
|
aicar
|
AB
|
T78L
|
1.1 / 0.8
|
10.9 / 2.2
|
-
|
|
|
AB
|
T78L+S101I
|
1.1 / 0.5
|
10.6 / 1.6
|
-
|
|
|
AB
|
R5A
|
1.0 / 0.8
|
11.0 / 1.9
|
-
|
The difference between the structures
seen from simulations :
channel in the aicar and glut structures
is more open than in gln and imgp+gln
aicar and glut structures have open
salt link tetrad for ammonium ion, which passes salt link 4 times more
often than neutral ammonia does. Aicar EF structure is different
from AB and CD (has different residues 23-24);
in the gln structure salt link tetrad
seems to be closed for ammonium. This correlates with the observation of
hydrogen bondng between OH_Tyr_B138 and NZ_Lys_A99 in the gln structure..
Yeast structure 1jvn has larger channeling
rate, because the starting point is more buried (but still accessible for
the probes ~ 0.5 A) .
Mutation influence is different for
different probes. Maximal influence of mutations are the rate
decrease by a factor of 1.4 forT78L (probe radius 0.4 A), 4.6 for T78L+S101I
(probe radius 0.5 A), 1.7 for R5A (probe radius 0.7 A)
Electrostatic
potentials at GLN and GLU binding sites
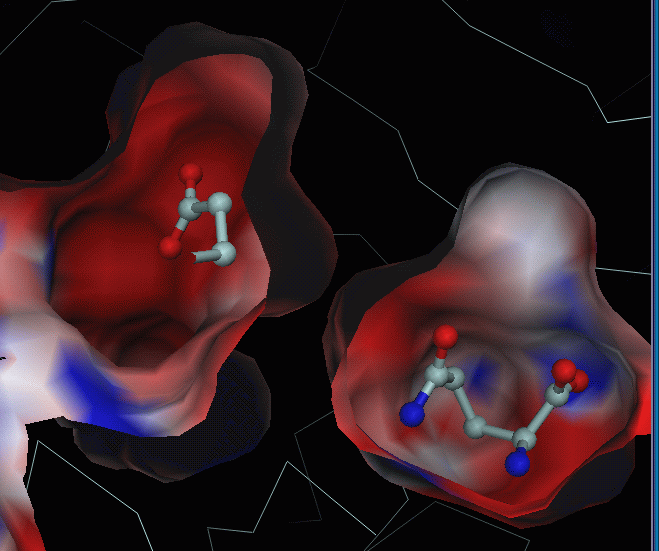
Below: electrostatic potential computed
for gln-AB structure and mapped onto the surface. GLN substrate (right)
is not used in calculations, but put back for visualisation purposes.
GLU substrate (left) is taken from glu-AB structure (superposed to gln-AB).
Below: electrostatic potential computed
for glu-AB structure and mapped onto the surface. GLU substrate is
not used in calculations, but put back for visualisation purposes.
GLN substrate is taken from gln-AB structure (superposed to glu-AB):
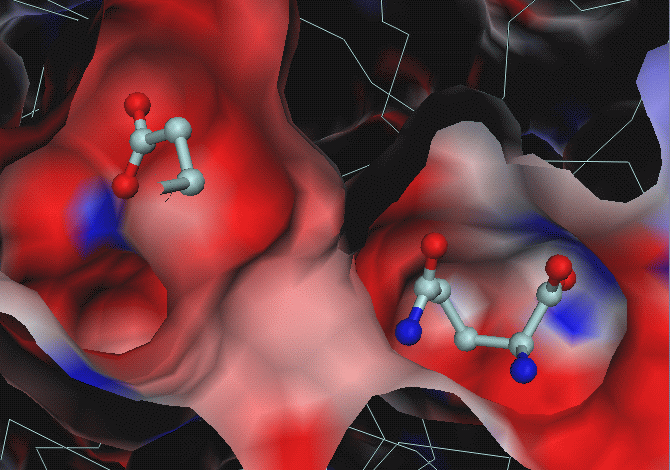
From these images it is not clear why GLU
is moved to its pocket, which is even more negative than GLN's pocket.
From hydrogen donor/acceptor pairs analysis
one can see that there is 1 hydrogen bond donor for GLU substrate in GLU
pocket: NE2_Gln_A123 (at 3.4 A from OXT of GLU) and one potential donor
ND1_His_B178 (at 3.43 A from OE2 of GLU, it is the only reason for blue
spot in the GLU binding pocket).
In the GLN pocket there are more donors
for GLU : N_Gly_B52 (for OE2), NE2_GlnB88 (for OXT), N_ThrB142 (for O),
N_Tyr_B143 (for OXT). I.e. from this viewpoint, GLN pocket is more
favourable for glutamate than GLU pocket.
I have done electrostatic binding free
energy calculations for GLU bound to HisHF. GLU bound to GLN pocket of
gln structure was modeled simply replacing NE2 to OE2 .
| Structure |
Chain |
Pocket |
Energy (van der Waals dielectric surface)
(kcal/mole) |
Energy (connoly dielectric surface) (kcal/mole) |
| gln |
AB |
GLN |
-3.6 |
+1.5 |
|
EF |
GLN |
-1.9 |
+7.3 |
| glut |
AB |
GLU |
+2.8 |
+22.0 |
|
CD |
GLU |
+3.7 |
+27.1 |
Again, with any dielectric surface treatment,
for glutamate, GLU pocket is less favourable than GLN pocket.
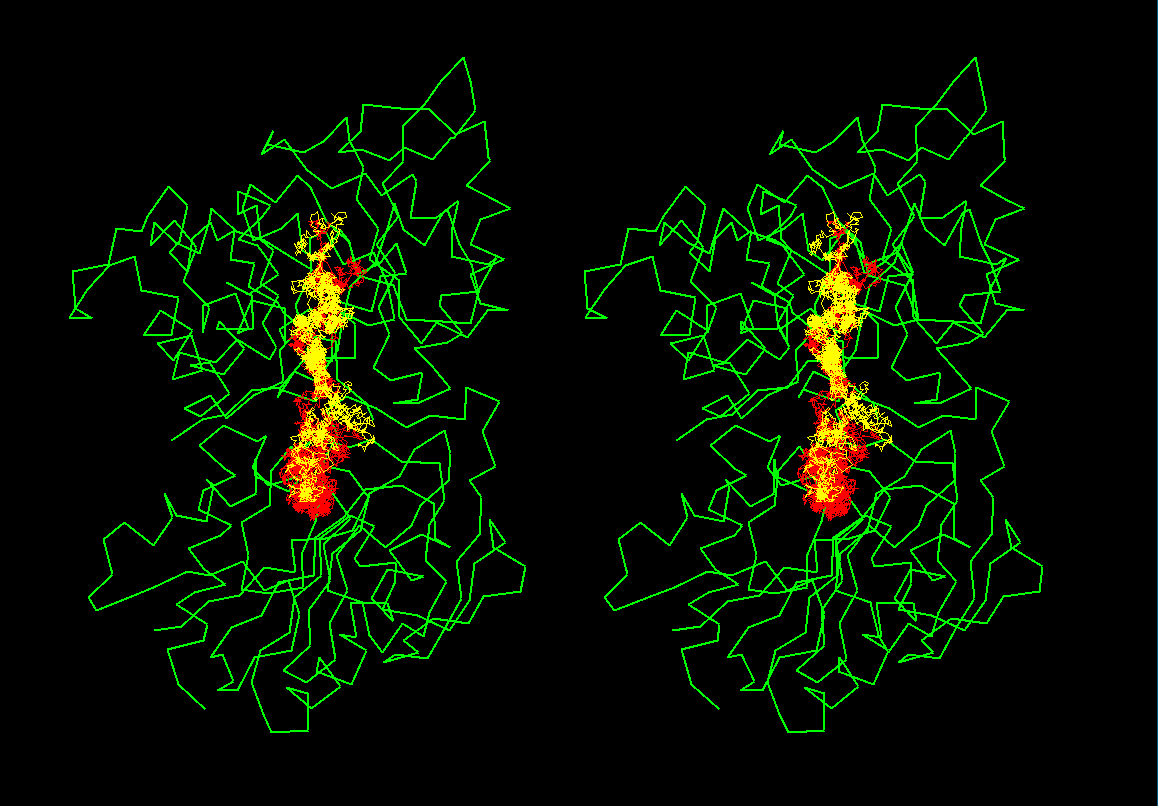
Brownian dynamics
simulation trajectories
There are 2 trajectories shown as yellow
and red taken from ca 1-2 ns long simulations. Trajectories start at the
lower part

R.Gabdoulline