Rotamerically Induced Perturbations with Langevin dynamics (L-RIP) & Rotamerically Induced Perturbations of a pseudo-Ligand (RIPlig)
L-RIP and RIPlig are two non-equilibrium MD approaches aimed at identification of slow conformational changes of a binding site
They are an extention of the Rotamerically Induced Perturbation (RIP) MD approach proposed by Ho and Agard (http://boscoh.com/rip/), which employs perturbation of side-chain torsional motion for initiating large-scale protein movement. RIP enables protein flexibility that normally occurs on the microsecond or longer time scale (for example, distortion of secondary and tertiary structure) to be explored.
In Langevin-RIP (L-RIP) approach perturbation is applied to the side chains of the binding site residues, but the original RIP procedure is modified by using a Langevin thermostat in the simulations instead of the original constant-energy conditions. This ensures that the average protein temperature is preserved during the MD simulations. Additionally, the friction term enables the elements of the protein structure with fast relaxation times to equilibrate within each perturbation pulse. This makes it possible to increase the number of pulses and, thus, enhance sampling of slow conformational variations.
In RIPlig, the perturbation procedure is applied not to each protein residue, but to an artificial molecule placed in the binding pocket with repetitions starting with different positions of the molecule, which enables flexible regions of the binding site to be identified in a small number of MD simulations of several picoseconds duration.
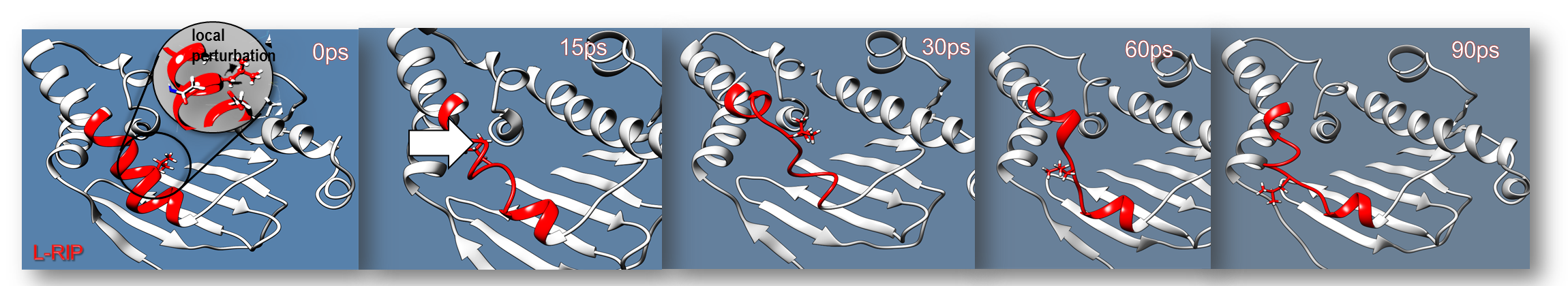
Running L-RIP or RIPlig
L-RIP & RIPlig are implemented as a part of RIP package written in Python and making use of NAMD software.
A modified package can be downloaded and executed as a stand-along program.
L-RIP & RIPlig methods are also implemented in a TRAPP web-server as a part of a workfolow for simulation of the binding site flexibility and prediction of transient binding pockets.
