RIP examples:
-
Documentation for the original RIP examples (to simulate the enkephalin peptide and perform flexibility analysis of a zinc-finger mimic) can be found at http://boscoh.com/rip/
L-RIP examples:
USAGE:-
The number of cores to be used can be defined in the corresponding ref.config file.
Then, the script can be executed as:
python CORRECT_PATH/pdbtool/perturb.py CORRECT_EXAMPLE_PATH/confg_file
For example, from the directory "enkephalin-NAMD_GBIS":
python ../../pdbtool/perturb.py enkephalin.config
-
The trajectory can be visualized in VMD (http://www.ks.uiuc.edu/Research/vmd/) using the command: "vmd md.psf md.dcd"
- Short test example: Enkephalin peptide
This is a short test example taken from the RIP package, but performing L-RIP simulations.
The input configuration file for L-RIP simulations, as well as the PDB file, can be found in the directory "examples/enkephalin-NAMD_GBIS".
To run the example, go to the directory "examples/enkephalin-NAMD_GBIS" and start the python script with:
'python ../../pdbtool/perturb.py enkephalin.config'
The simulations will only take a few minutes on a single processor.
The simulation results (MD trajectory for each perturbed residue placed in directories "300k-rip-300k/*/" and "10k-rip-26k/*/") can be visualized in vmd using the command:
'vmd md.psf md.dcd'
Because of the small size of the peptide, there is no large difference between simulations using L-RIP and RIP
More details on how to run an example and visualize results can be found here http://boscoh.com/rip/
- Comparison of RIP and L-RIP simulations: Distortion of the alpha-helix3 in the N-terminal domain of HSP90 (PDB 1UYD) upon perturbation of Leu107
The input configuration file for L-RIP simulations (100 pulses per 0.1ps and 300 pulses per 0.3ps) as well as the PDB file can be found in the directories
'examples/HSP90/LRIP-short' and 'examples/HSP90/LRIP-long', respectively.
The output trajectories are in:
'examples/HSP90/LRIP-short/300k-rip-300k/91' or 'examples/HSP90/LRIP-long/300k-rip-300k/91', respectively.
The simulations take about 30 minutes on 2 cores for L-RIP-short and 1(2.5) hours on 4(2) processors for LRIP-long.
Alpha-helix3 (blue) undergoes distortion in the middle, converting into two short helices.
The helical conformation is not observed in the apo-crystal structures, but provides an additional sub-pocket for ligand binding (crystal structures are shown below).
L-RIP: 100 pulses and
100 MD steps in each pulse
L-RIP: 300 pulses and
300 MD steps in each pulse
RIP: 100 pulses and
100 MD steps in each pulse
RIP: 300 pulses and
300 MD steps in each pulse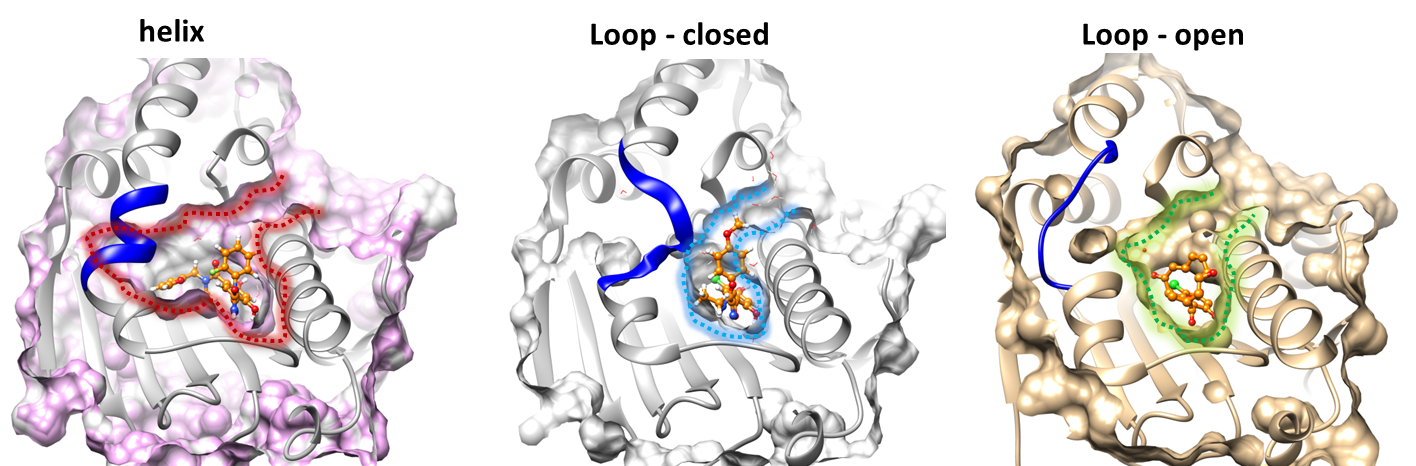
The movies demonstrate the perturbation of alpha-helix3 observed in short and long L-RIP as well as in short and long RIP simulations.
The perturbed residue is shown with carbon atoms coloured green.
The trajectory consists of the last snapshots from each pulse. Thus, the first frame of the trajectory already shows some perturbation.
Note, that the MD run in L-RIP uses NAMD and the CHARMM 2.7 force field, while RIP uses AMBER and the ff99 force field.
- To initiate micro-second timescale motion, long perturbation (0.3 ps per pulse) trajectories are required: Motion of the DFG loop in SRC tyrosine kinase (PDB 3U4W) upon perturbation of Phe405
The input configuration file for L-RIP simulations (300 pulses per 0.1ps) and an input PDB file is in the directory
'examples/SRC/LRIP' or
pregenerated trajectories can be found in:
'examples/SPR/LRIP/300k-rip-300k/91' or 'examples/HSP90/LRIP-long/300k-rip-300k/91', respectively.
DFG loop motion and flipping of the Phe405 are observed only wth long (0.3 ps) MD equilibration at each pulse. As the number of pulses increases from 100 to 300, multiple flips of Phe are observed, as well as larger oscillations of the beta-sheet (on the left of Phe405)
L-RIP: 100 pulses and
100 MD steps in each pulse
L-RIP: 100 pulses and
300 MD steps in each pulse
L-RIP: 300 pulses and
300 MD steps in each pulse
RIPlig examples:
- DFG loop motion and flipping of the Phe405 in SRC tyrosine kinase (PDB 3U4W)
In this example, the RIPlig trajectory is generated for only one position of the pseudoligand.
Each pulse consists of 100 steps of MD simulation
The input configuration file for RIPlig simulations and an input PDB file is in the directory 'examples/SRC/LRIP'.
Pregenerated trajectories can be found in: 'examples/SRC/RIPlig/RIPLIG4_PHE4/300k-rip-300k/276/'.
DFG loop motion and Phe405 motion are already observed after the first 100 pulses (0.1ps in each pulse); after 300 pulses, Phe405 has turned by about 180 Degrees, opening a transient sub-pocket behind it; simultaneously, the hinge motion of the two domains is initiated and the activation helix-loop unwinds (shown in orange).
100 MD steps (0.1ps) in each pulse
