The input file consists of a series of grouped input parameters, delimited by the keyword GROUP
GROUP = groupname
...
END GROUP
and a list of free parameters which are not associated with a group.
All input parameters have the following form :
name-of-parameter
= parameter-value
Lines begining with the character "#" or "!" are treated as
comment lines and will not be parsed by the program.
Note: do not insert spaces in front of "#" if you want to
comment out a line!
The tabs below contain different examples of input files. All parameter names are linked to a short summary. A full list and description of the parameters is given below the tabs.
-
computation [string] (off_rc) : define which type of simulation is
being performed. Options:
- association : compute association rate at every distance and for any number of contacts defined in group RateCalculation. Use *.rxna file and dind to define the criteria and their independence, all criteria are considered as nonspecific.
- docking : a complex must satisfy all specific criteria and at least nnnons non-specific ones in order to be saved in the complexes file
- electron_transfer : compute elctron transfer. The reaction criteria are read from 2 input files ( et_sol1 and et_sol2 )
- all : all complexes are saved to the complexes file. No checks are performed
- off_rc : disactivate the reaction criteria module, no calculations performed
-
rxna12f [string] (p12.rxna) : filename for the reaction criteria
used for docking or association rate
calculations.
Input examples for barnase-barstar docking and association rates are available in the examples. Reaction atoms do not have to be real atoms, although in this file these atoms/points should be given like atoms in PDB format. Different combinations of the same atoms/points can be prepared by editing these reaction atom files - et_sol1, et_sol2 [string] (empty) : filenames for the reaction criteria for electron transfer calculations.
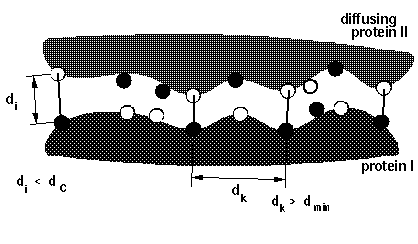
- dind [float] (6.) : minimal distance (in Å) between atoms on the same solute defining contacts for satisfying reaction criteria (equivalent to d_min in figure below)
- nnnons [integer] (2) : minimum number of nonspecific constraints to satisfy to define an encounter complex
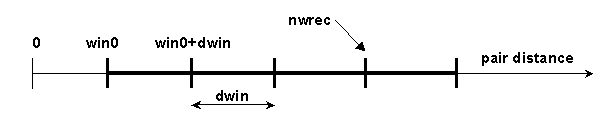
- nwrec [integer] (0) : number of the window up to which the complexes are recorded. For example, in the situation below, nwin=5 reaction distance windows are defined, and rates for forming contacts at each of these 5 distances will be monitored. Encounter complexes with contact distances less than win0+dwin*(nwrec-1), will be recorded.
- sdamd [integer] (0) : set equal to 1 to activate the option of recording encounter complexes at the windows distance according to win0+dwin*(nwrec-1). For example, in the case where win0=4.0, dwin=0.5, nwrec=23, encounter complexes will be recorded with at least one reaction contact at a distance of 15A. This option can be used in BD simulations of molecular association to generate encounter complexes for subsequent molecular dynamics simulations.
Reaction criteria are used to define a "complex" between 2 solutes. Full freedom is let to the user:
- Define a center-to-center distance criteria between the solutes, usually for docking
- A set of donor-acceptor pairs from an initial complex, needed for association rates
- Any other constraints suitable with experimental data: distance between 2 atoms satisfying crosslink experiments, FRAP or FRET data, known interactions with a specific residue / ligand...
- All constraints can be freely combined
The list of reaction criteria must be provided in a separate file (conventionally named with a suffix *.rxna), where the fields indicate:
- The keyword for the type of reaction criteria (discussed below)
- The position of the atom of the solute 1 (the atom number is needed in the case of a flexible solute)
- The minimum distance that the 2 reaction criteria must satisfy
- The position of the atom of the solute 2 (the atom number is needed in the case of a flexible solute)
For docking, there are 3 different types of reaction you can use:
- CSPEC: specific constraint, this reaction criterion must be satisfied for the complex to be accepted
- CNONS: nonspecific constraint, at least nnnons of these reaction criteria must be satisfied
- ASPEC: anti-specific, this criterion must be invalid (new in SDA 7)
For association rate calculations, all entries in the *.rxna file are considered as CNONS. But you can still record complexes and adjust the nwrec input parameter.
To get the specific format of the *.rxna file, we advise you to look at the input files in the barnase-barstar example of docking. Here are illustrated:
- A blind simulation, where only the center to center distance between the protein is used as a criterion
- An example where the center-to-center distance criterion is used together with nonspecific reaction criteria
(The doxygen documentation must be generated before using this link).
Usage of dind
To avoid an artificially large number of contacts, the
variable dind can be used.
On the figure below, all atoms used in the reaction
criteria are represented by spheres. But only those which
are at a separation greater than d_min are considered as
independent (black spheres). So with this schema, there
will be a maximum of 4 independent contacts.
Calculate bimolecular association rate constants using the Northrup, Allison and McCammon method (see Northrup, Allison McCammon, (1984), J. Chem. Phys.).
- win0 [float] (3.0) : reaction distance window minimum value (in Å)
- nwin [integer] (0) : number of reaction distance windows.
- dwin [float] (0.5) : reaction distance window step (in Å).
-
- nb_contact [integer] (4) : maximum number of pair contacts to be considered. This parameter is only applicable for association rate calculations.
- bootstrap [bool] (0) : if 1, association rates and first passage time results are printed for every trajectory. Final results can be computed with bootstraping (auxi/Bootstrap_multiCPU.py)
- fpt [bool] (0) if 1, record the first passage time, if bootstrap==1 same processing with (auxi/bootstrap.py)
- stop_traj [bool] (0) if 1, simulated trajectories will stop when the most strict reaction criteria definition is met (i.e. greatest number of contacts at the closest distance window). This may speed up your simulations, particularly in the case of strongly interacting simulations, although this will depend strongly on your choices of nb_contact and win0.
- analytical_correction [bool] (0) if 1, the analytical correction for centrosymmetric forces is computed for estimating the bimolecular association rate constant with the Northrup, Allison and McCammon method . When using this option, it is required that the interaction potential is centrosymmetric at the b surface, but it can be non-zero. This option requires that the real_net_charge and dh_radius is set for all solutes in the solute grid groups of the input file, and that the ionic_strength is set in the analytical group. If 0, there must be no interaction forces between the solutes at a separation of start_pos
- type [string] (sphere) : specify the geometry of the simulation
-
- sphere : : Use a spherical geometry, only for use with sda_2proteins types of simulation
- box : : Use a box for simulation, only for use with sda_2proteins types of simulation (protein-surface case) and with sdamm
- nambox : : Use a box for simulation and sphere to track distance of 2 solutes, for use in sdamm when calculating kon with NAM algorithm.
- pbc [bool] (0) : Switch on Periodic Boundary Conditions. Apply only with a box geometry
- surface [bool] (0) : Indicates if a surface is present in the system. PBC rules are modified in this case. Can also be used with a spherical geometry in sda_2proteins simulations
- escape [bool] (1) : Indicates if a solute can escape. The trajectory is then stopped when the solute reaches the c_surface (with a sphere) or zmax (in a box)
-
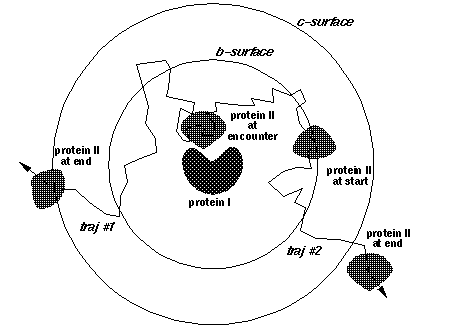
start_pos [float] (100.) : Indicates the initial position (in Å)
of the second solute in sda_2proteins. Simulations are
started by placing the center of the second solute at
the distance start_pos .
This corresponds to the b-surface for a spherical
geometry.
If surface is activated, the initial position is generated on the upper-half of the sphere.
For calculation of association constants, there should be no interaction between the solutes at this distance if analytical_correction = 0, or the interaction should be centrosymmetric if analytical_correction = 1. To ensure there are no grid-based interactions at this distance, start_position can be selected to be larger than the sum of the solute 1 (or 2) grid extents and the solute 2 (or 1) radius.
To check with box. - c [float] (150.) : defines the c-surface (in Å) in the case of a sphere
- <X>min,<X>max [float] (0.) : where <X> = x, y or z. Defines the size of the box (in Å). If escape is activated, zmax has the role of the c-surface.
- half_sphere [bool] (0) : Replace the spherical b surface with a spherical cap surface above an xy plane at a height z = min_height. This option is always used in the presence of a surface.
- min_height [float] (0.0) : defines the height of the spherical cap, relative to the centre of the centre solute, when option half_sphere is used.
Typical setup for simulations within a sphere. Trajectories are started at the b-surface (start_pos) and finish when solute 2 goes through the c-surface (c).
- variable [bool] (1) : switch to a variable timestep ( in sda_2proteins simulation), or to a fixed timestep (sdamm) when the value of dt1 is used
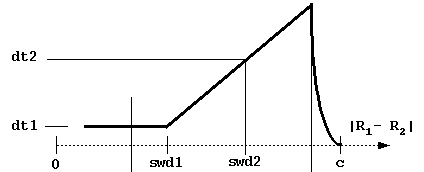
- dt1 [float] (1.0) : basal simulation timestep (in ps, picoseconds). This is the smallest timestep used and is employed when the center-to-center separation is less than swd1 or the distance at which the solutes have the possibility to contact (i.e. the center-to-center distance between solutes is less than rhit = the sum of maximal extents + probe radius + maximal radius of solute 1 atoms).
- swd1 [float] (50.0) : center-to-center distance (Å) at which the timestep starts to increase
- dt2 [float] (20.0) : timestep at distance swd2
- swd2 [float] (90.0) : center-to-center distance at which timestep equals dt2.
- height_dependent_dt [bool] (0) : if set to 1, the variable timestep depends on the z coordiate of the mobile solute, rather than the centre-to-centre distance between the solutes. This option is always used when a surface is present.
- rswd [*] (*) : deprecated in SDA 7, the rotation of solute2 is only proportional to the timestep
These parameters define the variable timestep designed to accelerate simulations. As shown in the figure below, the timestep is automatically decreased to zero near the c-surface to reduce artifacts due to truncation of the trajectories. Although the profile of the time step influences the computed rates, the most important parameter is dt1. Take care not to choose a too small value for this parameter (since the computing time scales almost linearly with it). The physical parameter sqrt(dt*dm) measures an average Brownian dynamics step length. This must be less than 1 Å, which is the characteristic distance describing the roughness of any protein surface since the atomic bond lengths and van der Waals radii define this scale. Moreover, dt1 should be less than the characteristic distance over which the interaction forces vary substantially, which means that the optimal value is influenced by interaction force strength: one needs smaller time steps for stronger interacting proteins. The standard output will print additional information, like the location and the value of the maximum timestep.
- This energy term has been parametrized for computing Au(111)-protein interaction energies. Computation of forces has not been parameterized:
- distance_to_surface [float] (6.5) : in Å (Zadw); the position of the first hydration layer (3 Å) plus the average LJ radius of protein-surface interaction
- energy_per_water [float] (0.13) : Φ°_metd in kT/Ų (Eadw); desolvation energy per unit area of the first hydration layer
- radius_patch [float] (6.0) : α in Å; (Aadw) parameter for calculation of the surface desolvation potential Φ as parametrized from MD simulations of PMF for the test atom on the Au(111) surface
- radius_water [float] (1.5) : Radw in Å; The effective desolvation radius that defines the surface area that each protein atom is assumed to desolvate.
The surface desolvation energy is calculated as :
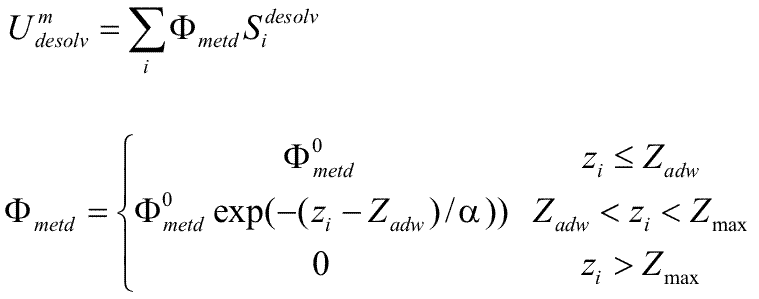
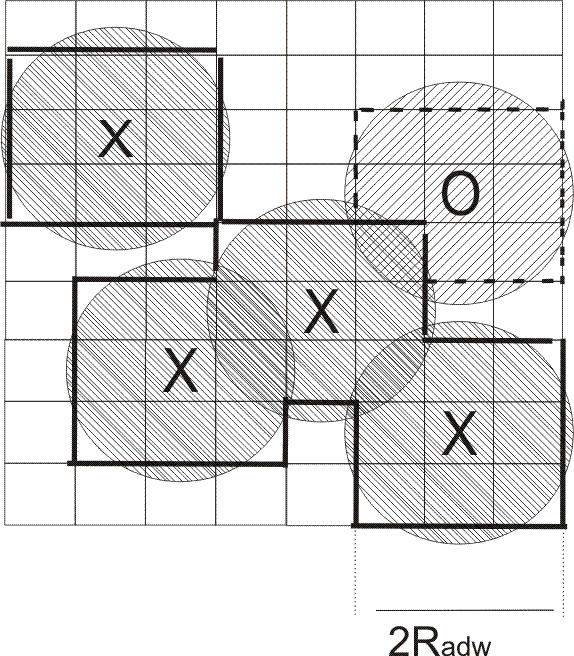
The computed desolvated area is shown by bold lines, the solid and dashed lines corresponding to water desorption from the first and second hydration layers, respectively. The circles with crosses represent close solute atoms and that with a zero is at an intermediate distance.
- Potential of mean force (PMF) calculations are based on a thermodynamic integration method where the configurational space of the solute with respect to a surface is sampled over the Euler angles (θ, ϕ, ω) and a number of dx,dy and dz coordinates based on the parameters:
- mentth [integer] (60) : Number of θ angles to be explored in rotational sampling. The angles range between 0 and π with a stepwise increment of (π ÷ (mentth - 1))
- mentfi [integer] (120) : Number of ϕ angles to be explored in rotational sampling. The angles range between 0 and 2π with a stepwise increment of (2π ÷ (mentfi - 1))
- mentom [integer] (60) : Number of ω angles to be explored in rotational sampling. The angles range between 0 and 2π with a stepwise increment of (2π ÷ (mentom - 1))
- mentx [integer] (6) : Number of subdivisions (dx) of the surface along the x axis to be sampled. The sampling length in the x direction is 6 Å
- menty [integer] (6) : Number of subdivisions (dy) of the surface along the y axis to be sampled. The sampling length in the y direction is 6 Å
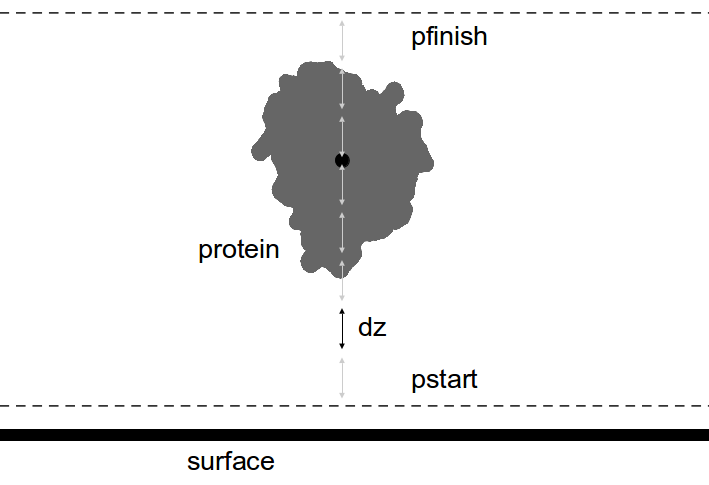
- pstart [float] (0.0) : Starting distance of the center of geometry of the solute from the surface in Å
- pfinish [float] (60.0) : Finishing distance of the center of geometry of the solute from the surface in Å
- dz [float] (0.2) : Increment step (dZ) along the z axis in Å
- dseed [float] (256.) : random number generator seed. If the value is 0, it uses the time clock. With OpenMP, it is not guaranteed that the same trajectory will be obtained with the same seed.
- nrun [integer] (1) : number of trajectories to generate. Only 1 is possible with SDAMM. The higher this number, the higher is the number of reactive trajectories (nrun_reactive). The relative error of the calculated rate constant is ~ 1/sqrt(nrun_reactive). This means that to have the same relative error, a larger value of nrun is required in cases with lower association rate constants.
-
timemax [float] (0.0) : maximum length of computed
trajectories in ps.
If timemax = 0.0, there is no limit; this is the recommended value for two-solute association rate calculations. - old_orientation [bool] (0) :Turn on the old version of computing the rotation matrix from the torque vector. In the new implementation, this conversion is performed using Rodrigues formula. However, in the old implementation, this conversion is performed by carrying out rotations first around the z-axis, then the y-axis and then the x-axis. This only works for infinitesimal rotations up to first order.
-
nprint : deprecated; frequency with
which rate of information is printed: the fraction of
reactive trajectories is printed every nprint
runs. This is most useful when the simulation
takes a long time, then intermediate results are
available after each nprint trajectories.
Values of the different probes
- probep [float] (1.7) : radius of the probe used to compute the exclusion volume grid. The surface of solute 1 that is excluded to solute 2 is defined by increasing the radii of the atoms of solute 1 that have a solvent accessibility greater than threshold by probep (Å). probep is usually chosen to have a value corresponding to the average radius of the surface atoms of solute 2, e.g. 1.77 Å for a protein. Typical values of this parameter are 1.4-1.9 Å. A smaller value (0.5 Å) should be used with the ProMetCS force field as it includes a Lennard-Jones term.
- probew [float] (1.4) : radius of solvent probe used to calculate the solvent accessibilities of solute atoms. A recommended value to use is 1.4 Å to represent a water molecule. This value is especially important if the hydrophobic desolvation term is used, because the calculated solvent accessibilities are used to calculate the hydrophobic desolvation energy and forces.
- threshold [float] (0.0) : solvent accessibility threshold - only the atoms of solutes with solvent accessible area greater than threshold (Ų) are designated as surface atoms. Moreover, only these atoms are used in calculating hydrophobic interactions and soft-core repulsion (repulsive Lennard-Jones). The default value is 0.0. This value is recommended for calculations with two solutes. A value of 5.0 is recommended for multiple solute (sdamm) simulations and for these, genbox, rather than SDA, should be used to compute the solvent accessibilities. The threshold value of 5.0 is used by genbox (with probew = 1.4 Å) to compute the solvent accessibility file for each solute. These files are used by SDA if save_access is set to 1.
- hexcl [float] (0.5) : spacing of the exclusion grid with which a solute is represented. Does not apply to SDAMM calculations where the exclusion grid is replaced by a soft-core repulsion. hexcl defines the accuracy with which the shape of the solute is described. Typical values are 0.5-1.0 Å for all-atom models of proteins. It needs to be consistent with the timestep used for BD moves - the spatial accuracy of the protein representation should be comparable to an average BD move in realistic simulations.
-
Saving data computed during the initialisation phase
The files will be saved in the directory where the pdb files are deposited.
save_exclusion [integer,0,1,2] (0) : if > 0, tries to read (and write) the exclusion grid from/into the disk. The grid can be loaded in VMD.- 0 : force recomputation of the exclusion grid during the initialisation
- 1 : save the grid in binary format
- 2 : save the grid in ascii format
- 0 : force recomputation of the accessible atoms during the initialisation
- 1 : save the data in ascii format
Multiplicative factors applied to the grids
SDA allows simulations with intersolute forces computed from different combinations of contributing terms that should be set by the user.
- ---For simulations of two solutes with excluded volumes and no intersolute forces, set all the following factors to zero. For simulations of multiple solutes that do not interact except through soft core repulsion, set all the following factors to zero except lj_rep_fct which should be set to 1.0.
- ---Set ljfct=1.0 if simulating a protein diffusing to a metal surface with the ProMetCS force field. Otherwise, ljfct should be set to 0.0.
- ---For simulating a single protein or peptide diffusing to a metal surface with the ProMetCS force field, all factors except lj_rep_fct should be set to non-zero values. Set epfct=0.5, edfct=3.34, hdfct=-0.0065, ljfct=1.0, see below.
- ---To compute the electrostatic interactions beyond the extent of the electrostatic potential grid of a solute molecule using the Debye-Hueckel model, set debyeh=1 (see GROUP = Analytical). This is advisable for calculations at low ionic strength or if the size of the electrostatic potential grid needs to be restricted.
- ---For computing bimolecular association rate constants for two proteins, set epfct=0.5 and set edfct and iostr in ed.in as described below. The other factors are usually set to 0.0.
- ---For docking two proteins to generate diffusional encounter complexes, set epfct=0.5 and set all other factors to 0.0. This setting was used for rigid-body docking subject to biochemical constraints by Motiejunas et al. Proteins, 2008, 71:1955-1969 Article link. The encounter complexes generated were refined by short molecular dynamics simulations, allowing for molecular flexibility, and the procedure was evaluated for a diverse set of protein pairs. This setting, using the electrostatic interaction term only, has also been used for rigid-body docking of proteins to solutes containing nucleic acids. An alternative that may be considered for docking molecular solutes, or a molecule and a surface, is to also compute the short-range electrostatic desolvation and hydrophobic desolvation terms, in addition to the electrostatic interaction term, by additionally setting edfct and hdfct to non-zero values, see below. This combination is particularly relevant for the docking of solutes that have weak electrostatic attraction. While it has been applied to a variety of molecular systems, it has not yet been systematically parameterized for general application to docking to generate diffusional encounter complexes (or to computing bimolecular association rate constants).
- ---For simulating the diffusion of multiple proteins, set epfct=0.5, edfct=0.36 (with electrostatic desolvation grids computed with iostr= 0 in ed.in), hdfct=-0.0013 (see below on modifying this value), and lj_rep_fct=1.0. The same settings should be applied for simulating the diffusion of multiple proteins in the presence of a non-metal surface.
- epfct [float] (0.0) : factor by which the electrostatic potential grid values read in are to be multiplied. Should be 0.5 for all calculations to compute the electrostatic interaction force if no modifications are needed. (This setting is equivalent to setting 1.0 in previous versions of SDA)
-
edfct [float] (0.0) : factor by which the electrostatic
desolvation potential grid values are to be multiplied to compute the electrostatic desolvation forces.
The value 1.0 corresponds to the uncorrected
electrostatic desolvation penalty for a charge in a
solvent due to the low dielectric cavity of a solute
treated as a collection of van der Waals spheres. More
appropriate values of this factor can be derived by comparing SDA
calculated energies of recorded complexes with energies
calculated with more accurate methods (by solving the
finite difference Poisson-Boltzmann equation, for
example). The accuracy with which the parameterization reproduces Poisson-Boltzmann energies and forces will vary depending on the solute properties, their proximity and the environmental conditions. One of two parameterizations of the electrostatic desolvation forces should be used:
(1) edfct=1.67 with solute interior dielectric constant set to 4 and the dielectric boundary between the solute interior and high dielectric solvent defined by the van der Waals surface of the solute. In this case, the electrostatic desolvation term is dependent on ionic strength and is computed with the same ionic strength (iostr) assigned in the grid preparation in ed.in as for the Poisson-Boltzmann electrostatic potential calculations. This parameterization was derived to reproduce Poisson-Boltzmann interaction energies of a set of protein-protein diffusional encounter complexes and used for the computation of protein-protein diffusional association rate constants, see: Gabdoulline, R. R.; Wade, R. C., J. Mol. Biol. 2001, 306, 1139 - 1155 Article link. N.B. For simulating a protein-metal surface system using the ProMetCS force field, set edfct to 2x1.67 =3.34 to ensure that the contribution to the electrostatic desolvation term with the image charge model is accounted for (this is equivalent to using edfct=1.67 between two standard solutes).
(2) edfct=0.36 with solute interior dielectric constant set to 4 and the dielectric boundary between the solute interior and high dielectric solvent defined by the van der Waals surface of the solute. In this case, the electrostatic desolvation term is not dependent on ionic strength and is computed with the ionic strength zeroed (iostr = 0) in the grid preparation in ed.in. Note that the electrostatic interaction term remains dependent on the ionic strength used in the Poisson-Boltzmann electrostatic potential calculations. This parameterization was derived to provide a simple protocol for computation of protein-protein electron transfer rates at different ionic strengths, see: Gabdoulline R.R., Wade. R.C. JACS, 2009, 131, 9230 Article link, and is employed in webSDA. In subsequent SDAMM multiple molecule studies, this parameterization has been used with the interior dielectric constant set to 2, see e.g. Mereghetti P.M., Gabdoulline R.R., Wade R.C. Biophys. J., 2010, 99:3782-3791. Article link. - hdfct [float] (0.0) : factor by which the hydrophobic desolvation potential grid values are to be multiplied to compute the short-range attractive nonpolar interaction forces. In the SDA program, hydrophobic desolvation energies are calculated by multiplying the solvent accessibilities of the atoms of one of the solutes by the value of the hydrophobic desolvation potential grid calculated for the other solute. The hydrophobic desolvation potential grid should be calculated with parameters assigned so that the sum of contributions from all accessible atoms gives the buried solvent accessible area, i.e. with a=3.1 Å, b=4.35 Å, and factor=0.5 in hd.in. The hdfct factor has units of kcal/mole/Å2 and converts buried area into hydrophobic desolvation energy. Realistic values for this factor based on analyses of buried surface area and known binding affinities of protein-protein complexes lie in the range range -0.0050 to -0.060 kcal/mole/Å2 (-5 to -60 cal/mole/Å2). In the study of plastocyanin-cytochrome F electron transfer, in which the hydrophobic desolvation forces were first introduced into SDA, the best agreement with experiment was obtained when hdfct was assigned a value of -0.019 kcal/mole/Å2. The more negative hdfct is, the more favorable the interaction between the solutes and the longer the residence times of the solute complexes: this can lead to the problem of the simulations becoming very computationally demanding. In this study, values of hdfct from -0.013 to -0.019 kcal/mole/Å2 were used, see: Gabdoulline R.R., Wade. R.C. JACS, 2009, 131, 9230 Article link. In subsequent publications describing simulations with hydrophobic desolvation forces, values in the range from -0.005 to -0.019 kcal/mole/Å2 have been used, with most studies done with hdfct = -0.013 kcal/mole/Å2. However, note that for simulations of protein-gold surface interactions with the ProMetCS force field (which also includes metal desolvation and Lennard-Jones terms), hdfct = -0.005 to -0.0065 kcal/mole/Å2 is used, see: Kokh et al. JCTC, 2010, 6, 1753 Article link.
- ljfct [float] ( 0.0) : factor by which the Lennard-Jones potential grid values are to be multiplied when using the ProMetCS force field.
- lj_rep_fct [float] ( 0.0) : factor by which the soft-core repulsion (repulsive Lennard-Jones) grids are multiplied to compute the soft-core repulsion forces. It was introduced for use with sdamm for multiple molecule simulations, and replaces the use of the exclusion grids. If the repulsive Lennard-Jones grids are computed with an assignment of factor=4096 in ljrep.in, then lj_rep_fct=0.0156 should be used (see also the Preparation tools documentation).
- oneway_surf_charge [bool] (0) : if set to 1, the electrostatic interactions between a surface and a solute will be computed in one direction with the grid for the surface interacting with the (effective) charges on the solute. If set to 0 (default), the electrostatic interactions are computed in both directions (average of surface grid with solute charges and grid charges with solute grid)
- epfct_oneway_surf [float] (1.0) : scaling factor for the one-way electrostatics which should be set to 1.0 (default).
-
Other parameters
- ignore_exclusion [bool] ( 0 ) : if set to 1, the excluded volume check will not be performed to prevent solute overlaps. In this case, soft-core repulsion should be used instead.
- restart [string] ( empty ) : input file from which the initial solute positions are read (sda_energy, sda_koff, sdamm). If this is commented, the positions in the input pdb files will be used as the starting positions instead (for sda_koff)
- account_occurency [bool] ( 0 ) : Only in the case of sda_koff, if set to 1, the trajectory will restart n_occurency * nrun times with the initial position given by the complex.
- novers [integer] ( 150 ) : number of overlaps allowed before making a boost. Recommended value: 100-200. With a value in this range, boosts are usually not necessary unless the interactions are very strong. This can be the case if a very small probe size (probep) is chosen. Many boosts indicate erroneous input.
- rboost [float]( 1.0 ) : boost distance (in Å) to move the solutes apart when more than novers moves were not accepted due to overlaps. Boosting is introduced to avoid infinite trajectories and can influence simulation results because trajectories are modified by boosts. The number of boosts is reported in the output log.
-
Parameters specific to the ProMetCS force field
-
correction_image_charge
[float]( 0.0 ) : a non-zero positive number means that
the analytical correction will be applied in computing
the image charge electrostatics. This parameter is also
used to re-scale effective charges when image-charge
electrostatics is computed analytically (at small
distances from the surface when the solute-surface
interface is partially desolvated).
If the protein effective charges do not deviate strongly from the test charges in magnitude (this is usually the case for proteins), use correction_image_charge = 1.
However, if the effective charges of a solute are too small (as for the case of DNA simulation), the parameter correction_image_charge enables correcting back (particularly, to increase) solute charges to test charges when they are used for the calculation of image-charge electrostatics at small solute-surface distances
Note, that the parameter image_charge = 1 must be defined to use this function. -
If correction_image_charge > 0, the dielectric constant for the case of atomic cavities in the solute molecule close to the surface is scaled as follows:
ε=4.0+A*z*z+exp(-z/B - C) for 2.5 < z < 5.5 Å, where :- A ; default value: A = 0.8 1/Ų
- B ; default value: B = 0.39 1/Å
- C ; default value: C = 10.4
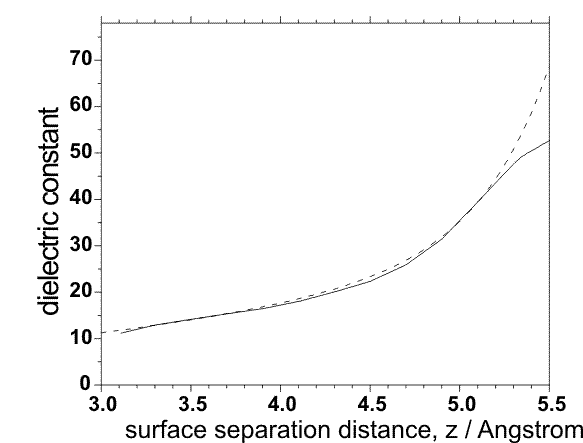
These parameters have been derived from MD simulations to compute the total electrostatic interaction energy for a test charge atom as a function of the distance from a gold surface and are hard-coded in the file mod_compute_image_charge.f90 (ep_scaleA, ep_scaleB and ep_scaleC)
-
desolv_image_charge
[integer](
0 ) : indicates if
the image charge calculation includes the electrostatic
desolvation term.
If it is equal to 0, it is deactivated but will be automatically switched on if the electrostatic image charge is computed.
To force the deactivation of this term in this case, use desolv_image_charge = -1.
Imprint/Privacy