
This example shows the implementation of the ProMetCS force-field in SDA 7.
It is parameterized for protein-metal surface interaction, including electrostatic image charge and metal desolvation energy terms. The ProMetCS force field is described in : Kokh et al.ProMetCS: An Atomistic Force Field for Modeling Protein−Metal Surface Interactions in a Continuum Aqueous Solvent.J. Chem. Theory Comput., (2010) 6:1753-1768.i DOI: 10.1021/ct100086j
It has been implemented in SDA 7 and is available for multiple proteins simulations (SDAMM). It is still under development.
We advise to compare your results with the SDA 6 released version for reliable results.
This example demonstrates calculations for (i) the diffusional docking of barstar (BS) to an Au(111) surfacei and (ii) the simulation of the diffusion of multiple BS molecules in the presence of an Au(111) surface.
This example can be found in directory examples/aubs.
| prepare_grids_and_ecm/ | Directory for generating grids and effective charges needed for running the simulation |
| data_grid/ | Data files (pdb and input files) needed for running the examples |
| run_hits/ | Results of the example run script for protein-metal surface docking |
| run/ | Directory to run the protein-metal surface docking |
| run_sdamm_hits/ | Results of the example run script for a multiple protein simulation |
| run_sdamm/ | Directory to run the multiple protein simulation |
| doc/ | This documentation |
| unit-test/ | For developers, regression tests for many different combinations of interactions. The grids need be generated first. |
The different components of the ProMetCS force field have been implemented in SDA 7 with the possibility to be selected individually:
- Electrostatic interaction with a metal surface: the electrostatic image-charge method is applied.
It is a term which can be applied with any conductor surface.
It must be activated for each type of solutes in the sda input file (image_charge = 1). - At short distance an analytic correction, modifying the dielectric constant close to the surface,
can be activated globally, for all solutes (
correction_image_charge = 1), at the exception of the surface. - The metal desolvation term, accounting for the desorption of the water molecules, can be adjusted with the input parameters
in the group
MetalDesolvationof the input file. It applies to all solutes, except the surface. - The electrostatic desolvation image-charge is implemented in a similar way to the electrostatic image-charge,
but with the electrostatic desolvation grid and the square of the effective charges.
It is activated by a global input parameter (desolv_image_charge = 1) and applies to all solutes except the surface.
Assure you have compiled all the executables and tools in sda_flex/bin/ first or refer to the compilation section.
Run first the script run-aubs_ed_hd_ecm_lj.sh from the prepare_grids_and_ecm directory to generate the grids necessary for this example
Note : the generation of the Lennard-Jones grids need more time than the other grids (approx. 1-2 hours)
Then go in one of the directory containing input file (run or run_sdamm) and run the shell script
For the protein-surface simulation :
cd run/
./run_sda_prometcs.sh
and for the multiple proteins-surface simulation :
cd run_sdamm/
./run_sdamm_prometcs.sh
You can check that output files are similar to the ones generated in the *_hits directory.
For visualization and analysis of the docking configurations of BS on the gold surface, a clustering procedure can be used.
For this one has to eliminate rotational (around Z) and translation (in (X,Y) plane) degrees of freedom of BS, since the interaction with the gold surface depends practically only on the Z coordinate (scripts DelRotZ_complexes-SDA7.py).
Then one can use either a k-means clustering (as in the example case bnbs) or a hierarchical clustering procedure (python code myClustering_complexes-SDA7.py, see script run_sda_prometcs.sh). The latter method enables clustering of the docking orientations with a pre-defined RMSD (5 Angsrom, for C_alpha) within each cluster.
| (cluster #) | (average energy) | (population) |
|---|---|---|
| 1 | -4.149e+01 | 2.419490e+05 |
| 2 | -3.030e+01 | 7.127600e+04 |
| 3 | -3.880e+01 | 7.419120e+05 |
| 4 | -3.219e+01 | 8.825750e+05 |
(Note that using a smaller number of low-energy complexes for clustering (for example, only 500), leads to fewer clusters (6 in the case of the 500 complexes analyzed). Nevertheless, the first 3-4 low-energy clusters will be preserved, though the average energy, population and order may be slightly different. (These clusters also correspond to the first three low energy clusters obtained in similar simulations with the SDA6).
| (cluster #) | (total energy) | (Lennard-Jones + Metal desolvation; kT) | (Image-charge + electrostatic desolvation; kT) |
|---|---|---|---|
| 1 | -44.0 | -3.9 | -37 |
| 2 | -42.5 | -5.8 | -35 |
| 3 | -42.2 | -36.7 | -1 |
| 4 | -34.6 | -15 | -17 |
The docking positions of BS on gold for each cluster were generated by trajectory2dcd:
| cluster #1 & #2 | cluster #3 | cluster #4 |
|---|---|---|
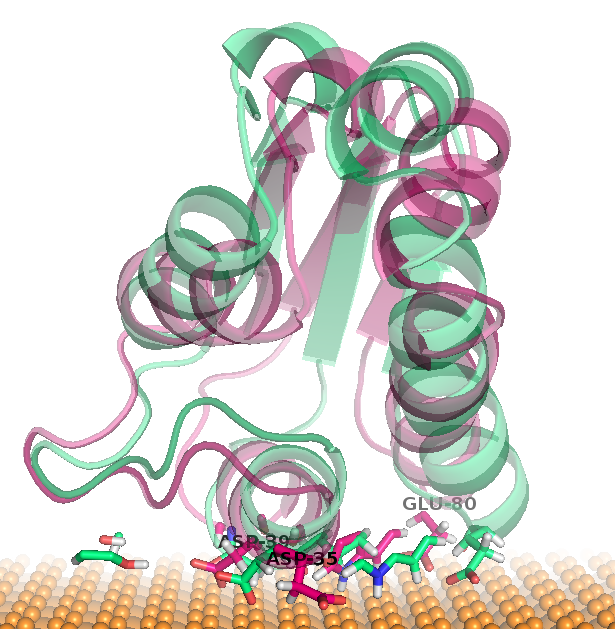
| 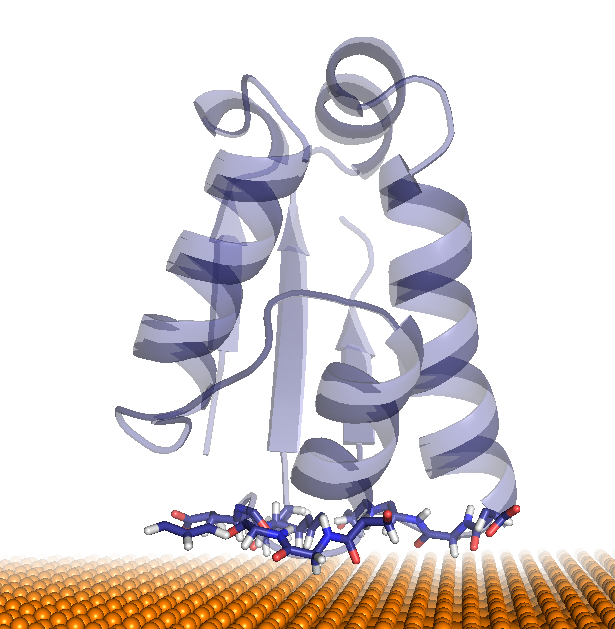
| 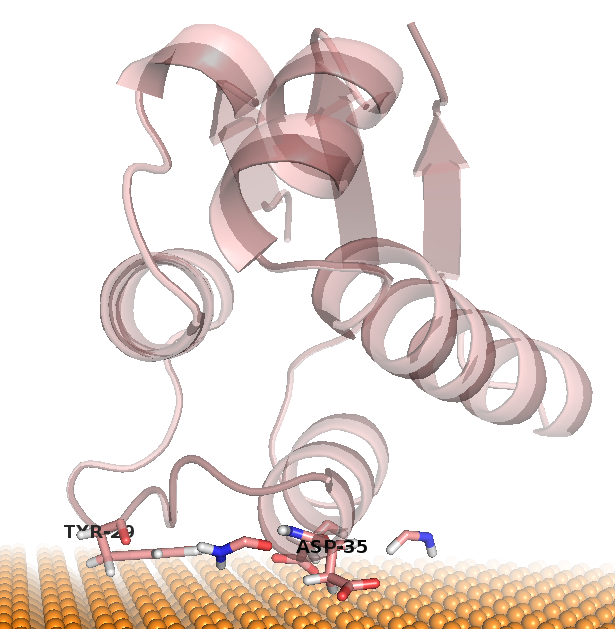
|
The extension of SDA 7 to multiple protein simulations in the presence of a gold surface is currently under development. The example demonstrates how simulations of this system can be run but the correct parameterization for protein-gold and protein-protein interactions cannot currently be specified in the sda input.
This type of simulation allows different arrangments of the solutes with the surface to be studied
at a high protein concentration.
Snapshot after 100 ns of simulation. 2 solute barstar molecules are bound to the surface in configurations similar to clusters #2 and #7 from the simulations with one barstar molecule
