
This script uses all pdb structures and grids in data_grid/. You need to run at least one time the script in prepare_grid_and_ecm/ to generate the grids and effective charges in this folder.
If you want more information about this step, refer to the tutorial in examples/Tutorial_SDA7/.
cd bnbs_docking/
./script_docking.sh
You can check that the output files are similar to the ones generated at HITS in bnbs_docking_hits/
Note: the script run_clusterDPipe.sh will be executed by the main script, but feel free to use it as a template.
The script will generate the most favorable docking positions between barnase and barstar in 2 ways:
- A "blind" simulation without any a priori constraints other than the requirement that a docked complex should have a protein center-to-center distance of less than 48 Å. Only electrostatic interactions are taken into account.
- The residence time in UHBD grid format,
resid3d.bin.grd, can be visualized in VMD. - A second simulation where, in addition to the center to center distance criterion, it is required that at least 2 hydrogen-bond donor-acceptor pairs are within less than 6 Å to record a complex. Additionally, the electrostatic desolvation and the non-polar terms are included in this simulation.
In both cases, the complexes saved during the SDA runs are subsequently clustered and 5 representative configurations are generated.
The results of the clustering are in their respective subfolders, in the file out_clustering.
You can find all the information about these outputs in the documentation
of the cluster program.
In both simulations, the first clusters are the most populated, with more than half of the complexes.
Furthermore, they have the lowest interaction energy, and the minimum RMSD (computed from p2_noh.pdb, which is the crystal structure).
The figure below shows the crystal structure of the barnase-barstar complex, and the representatives of the first clusters for the 2 simulations.
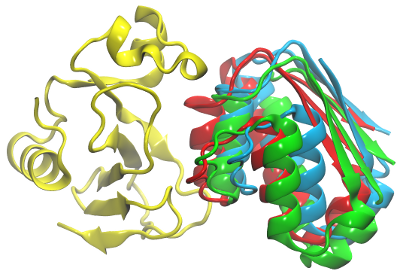
Red: barstar (solute 2) - position in the crystal structure of the complex
Blue: Representive from simulations with only the electrostatic interaction (RMSD 6 Å)
Green: Representative from simulations with all energy terms (RMSD 5 Å)